



Cuando queremos escribir una carta o firmar un documento importante solemos hacerlo con un bolígrafo de tinta, de modo que el texto contenga la impronta de la escritura personal. Algo similar sucede en el interior de nuestras células, cuya ‘tinta genética’ dicta la manera como los cuerpos van a funcionar. Cuando esa tinta se ‘entrecorta o difumina’ aparecen alteraciones en el ADN que pueden llevar a la enfermedad.
Tal es el caso de la mucopolisacaridosis iva (MPS IVA) y la enfermedad de Tay-Sachs (TSD, por su sigla en inglés), dolencias de origen genético cuyo ‘error de escritura’ genera (en cada caso) la falta de una enzima. Estas ausencias conducen a la acumulación de compuestos, dentro del lisosoma –encargado de mantener la célula limpia y reciclar sus componentes–, que no pueden ser degradados ni eliminados. Dicha carga provoca la muerte de las células y, con ello, efectos negativos en el cuerpo.
Al tratarse de enfermedades tan particulares, se requieren estrategias terapéuticas precisas. Ese es el enfoque que siguen los profesores Ángela Espejo y Carlos Javier Alméciga, investigadores del Instituto de Errores Innatos del Metabolismo (IEIM) de la Pontificia Universidad Javeriana, y cuyo trabajo va más allá de tratar los síntomas: ellos buscan corregir el error genético de enfermedades de base lisosomal con estrategias de edición genética.
“Este tipo de enfermedades lisosomales tienen esa cosa bonita, y es que puedes modificar una célula que producirá la proteína para ella misma, pero la que le sobre la compartirá con las vecinas, y es lo que se llama el mecanismo de corrección cruzada”.
Carlos Javier Alméciga Díaz
Un editor de tinta genética
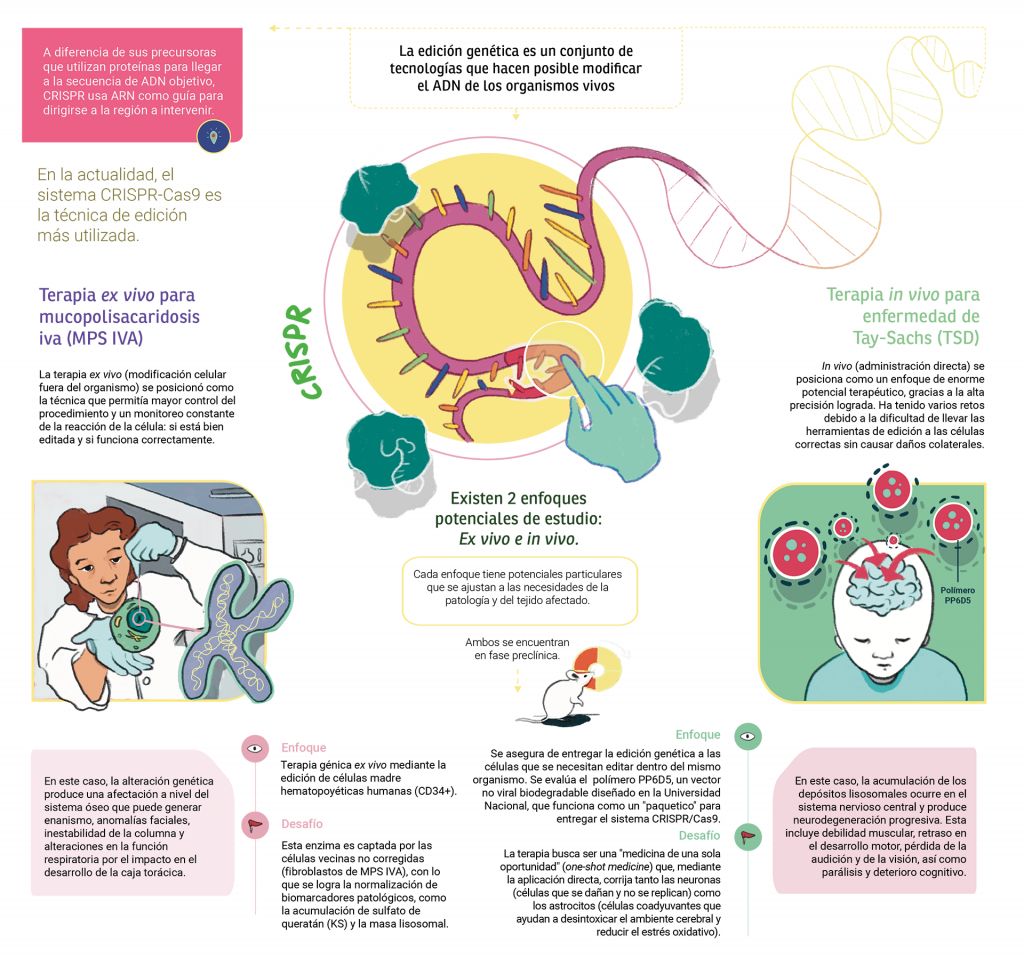
La edición genética es un conjunto de tecnologías que permite intervenir el ADN de los organismos vivos. Al editar, como en un procesador de texto, la ‘tinta genética’ inserta, retira o cambia fragmentos de la secuencia de ADN de las células, lo que permite que estas se reparen y corrijan el defecto que causa la enfermedad.Son varias las tecnologías que editan una secuencia genética. Entre las primeras en aparecer se encuentran las nucleasas de dedos de zinc y las nucleasas efectoras tipo activador de la transcripción.
Ambas usan enzimas (nucleasas) para intervenir el ADN en el lugar preciso y aprovechan la capacidad natural de la célula de reparar la hebra genética y poder insertar los cambios de escritura necesarios. Estas técnicas usan proteínas diseñadas artificialmente para lograr una edición dirigida, un proceso complejo que puede ser poco eficiente y tener efectos fuera de la secuencia objetivo.
Posteriormente aparece CRISPR-Cas9, que en la actualidad es la técnica de edición más utilizada debido a su versatilidad y eficiencia. A diferencia de sus precursoras que utilizan proteínas para llegar a la secuencia de ADN objetivo, CRISPR usa ARN como guía para dirigirse hacia la región a intervenir.
En lugar de enzimas diseñadas, esta tecnología emplea una sola proteína (Cas9) para separar las cadenas de ADN en el punto preciso. Su funcionamiento se basa en el sistema inmunológico natural de las bacterias para combatir infecciones. Al adaptar este mecanismo, fue posible crear una herramienta precisa y sencilla en comparación con sus antecesoras.
La manera como se ha implementado la edición genética ha cambiado a lo largo del tiempo y con ella la forma de orientar las estrategias que buscan resolver esos ‘errores sintácticos’. Por ejemplo, el enfoque ex vivo fue el primero en utilizarse, pues ofrecía un entorno más controlado al extraer las células del paciente para modificarlas en el laboratorio antes de reintroducirlas. Con esta técnica fue posible obtener mejoras en los síntomas asociados a diferentes enfermedades.
Por su parte, el enfoque in vivo ha tenido varios retos debido a la dificultad de llevar las herramientas de edición a las células correctas sin causar daños colaterales en los pacientes. Aunque hoy se utilizan ambos enfoques, la edición in vivo se posiciona como un enfoque de enorme potencial terapéutico, gracias en parte a la precisión lograda por CRISPR.
Gracias al recorrido realizado por la edición genética, hoy es posible avanzar hacia el desarrollo de tratamientos que logren editar la ‘tinta genética’, cuyos errores de escritura son responsables de enfermedades como MPS IVA o TSD. Esto permite generar tratamientos para cada enfermedad, de modo que la terapia genética llegue a las células específicas con un alto grado de precisión, explica el profesor Alméciga.
Curar células en el laboratorio y regresarlas al cuerpo
En el escenario de la MPS IVA, la alteración genética produce una afectación a nivel del sistema óseo que puede generar baja estatura, inestabilidad de la columna, enfermedad cardiaca y alteraciones en la función respiratoria, por el impacto en el desarrollo de la caja torácica. Para este caso, Espejo y Alméciga, en un trabajo colaborativo con investigadores del Nemours Children’s Hospital, en Delaware (Estados Unidos), exploran estrategias de edición genética ex vivo que contribuyen al manejo de esta enfermedad.
Esta técnica, con precedentes clínicos validados en otras enfermedades, permite editar en el laboratorio células que portan la mutación y que han sido extraídas del paciente. Allí, se monitorea la reacción de la célula: si está bien editada y si funciona correctamente. Luego de esta verificación, “se le inyecta al paciente, lo que aumenta la seguridad de la terapia versus si se administra a todo el organismo antes de explorar su resultado”, comenta el investigador javeriano.
En la MPS IVA, esta estrategia tiene un gran potencial debido a que se necesitan corregir varios tipos de tejidos, y al integrar células madre que ya han sido editadas se garantiza la seguridad para el paciente y la duración de la terapia a largo plazo. En todo caso, algo importante en este escenario de la edición genética “es que no todas las células quedan corregidas”, comenta el profesor. Sin embargo, “pasa algo muy bonito”, continúa, pues la célula modificada produce proteína para ella misma y “la que sobra la comparte con las vecinas”. Este fenómeno, conocido como corrección cruzada, extiende los efectos terapéuticos de la edición a células que mantienen el defecto genético.
El trabajo desarrollado por los investigadores y sus aliados en Delaware demuestra el potencial terapéutico del enfoque ex vivo al editar células madre hematopoyéticas humanas mediante el sistema CRISPR-Cas9. Los investigadores lograron que las células editadas mantuvieran su capacidad de especializarse y cumplir su función. Esto es clave para asegurar beneficios terapéuticos y continuar el camino de los estudios preclínicos en ratones para evaluar la seguridad de la terapia y avanzar hacia la fase clínica, de modo que se pueda tratar a pacientes humanos.
Un elegante sistema de distribución terapéutica
En la TSD, la acumulación de los depósitos lisosomales ocurre en el sistema nervioso central y produce neurodegeneración progresiva. Esta incluye debilidad muscular, retraso en el desarrollo motor, pérdida de la audición y la visión, así como parálisis y deterioro cognitivo. Actualmente, no hay un tratamiento para esta condición y una limitación importante tiene que ver con la barrera hematoencefálica, es decir, la capa protectora y semipermeable entre la sangre y el cerebro que recubre el sistema nervioso y actúan como filtro, la cual previene que sustancias nocivas y un gran número de moléculas terapéuticas puedan afectar este órgano.
Para resolver esta limitación, los investigadores del IEIM trabajan en un estudio in vitro que busca evaluar un polímero basado en nanotecnología con el que crean una especie de paquetes, donde introducen la terapia de CRISPR-Cas9, para entregarlos a las células. El polímero PP6D5 como estrategia de entrega no viral, se distancia de los vectores virales al ser más seguro: genera menos respuesta inmune, reduce el riesgo de mutaciones y puede transportar una mayor variedad de material genético. Esto lo convierte en “el mejor empaque para llegar de manera más efectiva a la célula”, complementa la profesora y doctora en ciencias biológicas.
El estudio concluye que PP6D5 es un material eficiente para entregar CRISPR-Cas9 a las células del sistema nervioso en el contexto de la TSD, en comparación con otros vectores no virales comerciales, cualidad que lo posiciona como una herramienta con alto potencial. Además, al ser biodegradable, no se acumula en la célula y permite maximizar la distribución en el tejido cerebral, añade la profesora Espejo.
Además, se trata de “un desarrollo colombiano”, complementa Alméciga. PP6D5 fue creado y diseñado por Ivonne Díaz y León Pérez, investigadores del Departamento de Química de la Universidad Nacional de Colombia. Estas colaboraciones entre equipos de investigación fortalecen las capacidades locales para la creación de terapias genéticas en el país.
El futuro de la genética es personalizado
Al reflexionar sobre los hallazgos de ambos estudios, el profesor Alméciga comenta que avances investigativos como estos constituyen una expansión de los trabajos en edición genética que vienen realizando como grupo. Este crecimiento busca abrir más posibilidades tanto en los sistemas de entrega como en el diseño de estrategias adaptadas a diferentes escenarios terapéuticos y a diferentes pacientes.
Este camino ha sido posible gracias a la financiación estratégica, principalmente por convocatorias internas de la Pontificia Universidad Javeriana, y al compromiso con el desarrollo local. Como explica Alméciga, parte de los fondos provienen de un círculo virtuoso generado por la transferencia de tecnologías desarrolladas en el IEIM al Hospital Universitario San Ignacio, y cuyos réditos se reinvierten en nueva investigación.
Con la mejora en el desarrollo de terapias genéticas a lo largo de varios años que se ha logrado en el IEIM, se abre la posibilidad de explorar otras enfermedades que requieren analizar de manera integral las afecciones y las características de cada paciente. Esta expansión del trabajo acerca al grupo a una medicina personalizada, que hará posible contar con un editor particular para cada línea de “tinta genética” que dibuje nuestros genes, pues, como explica Alméciga, “si solo tienes una oportunidad, tienes que aprovecharla muy bien”.
Para leer más:
§ Herreno-Pachón, A. M., Leal, A. F., Khan, S., Alméciga-Díaz, C. J. y Tomatsu, S. (2025). CRISPR/Cas9-edited CD34+ cells rescue mucopolysaccharidosis IVA fibroblasts phenotype. International Journal of Molecular Sciences, 26(9), 4334. https://doi.org/10.3390/ijms26094334
§ Guerrero-Vargas, J. M. et al. (2025). Evaluation of the PP6D5 polymer as a novel non-viral vector in the development of a CRISPR/Cas9-based gene therapy for Tay–Sachs disease. Pharmaceutics, 17(5), 628. https://doi.org/10.3390/pharmaceutics17050628
TÍTULO DE LA INVESTIGACIÓN:
Evaluación del efecto terapéutico del sistema de edición génica CRISPR/Cas9 acoplado a magnetoliposomas en un modelo animal de la mucopolisacaridosis IVA.
INVESTIGADOR PRINCIPAL:
Carlos Javier Alméciga Díaz
COINVESTIGADORES:
Ángela J. Espejo-Mojica
Instituto de Errores Innatos del Metabolismo
Facultad de Ciencias
Pontificia Universidad Javeriana
PERIODO DE LA INVESTIGACIÓN: 2022-2025
